
Tak jak każdy człowiek, tak i dziecko z zespołem Cri du Chat, dziedziczy wiele cech po swoich rodzicach i przodkach. Posiada także wiele niepowtarzalnych, indywidualnych cech. Są one wytworem swoistej gry wpływów czynników natury genetycznej i wpływu środowiska, które te cechy kształtują. Swoistą grę zależności genów od środowiska i odwrotnie, modyfikuje pojawienie się dodatkowo utraty materiału genetycznego w obrębie ramion krótkich chromosomu 5, co czyni je podobnymi do innych dzieci z tym zespołem. Ta modyfikacja sprawia, że obserwujemy wspólne cechy dla osób z zespołem Cri du Chat i ta wspólnota cech często przekreśla indywidualne widzenie dziecka, któremu to się przytrafiło.
Modyfikujące działanie niedoboru materiału genetycznego chromosomu 5p na fenotyp wyraża się wykształceniem szeregu cech wspólnych w wyglądzie twarzy, rąk, czy sylwetki ciała i odmiennym tempie rozwoju cech umysłowych. Charakterystyczne cechy twarzy mogą wskazywać na współistnienie wad rozwojowych narządów wewnętrznych, które należy ocenić, zanim powstaną istotne zaburzenia kliniczne. Cechy te tworzą spektrum cech, które upoważnia do postawienia rozpoznania i skierowania na badanie cytogenetyczne, aby zweryfikować diagnozę.
DNA, obecne w jądrze każdej komórki oraz będące nośnikiem dorobku genetycznego, podczas rozmnażania się komórek dzieli się na segmenty zwane chromosomami. Człowiek posiada 23 pary chromosomów. Każda para składa się z chromosomu pochodzącego od ojca i z chromosomu od matki. Para chromosomów, która określa płeć (chromosomy płciowe) to XX u kobiet i XY u mężczyzn. Pozostałe pary oznaczone są numerami od 1 do 22.
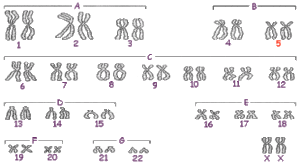
W przypadku syndromu Cri du Chat stwierdzono brak (delecję) fragmentu krótkiego ramienia jednego z chromosomów 5 (5p-) .
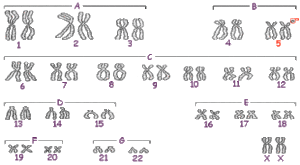
Delecja może dotyczyć części końcowej bądź wewnętrznej krótszego ramienia. W niektórych przypadkach jest ona spowodowana translokacją (przeniesieniem segmentu z jednego chromosomu na drugi), rzadziej może pochodzić z innych zmian chromosomowych jak na przykład: mozaicyzm, inwersja chromosomowa, chromosom pierścieniowy.
W większości przypadków delecja pochodzi z mutacji „de novo”, gdzie kariotyp obojga rodziców był prawidłowy. W 10 – 15% przypadków jedno z rodziców jest nosicielem aberracji chromosomowej (mutacji, która może być obecna u większej ilości pokoleń tejże rodziny), częściej jednak występuje translokacja, która nie zmienia swojego dorobku genetycznego (tzw. translokacja zrównoważona), ale która może spowodować delecję u dziecka. Przypadki te są nieprzewidywalne, a syndrom występuje bez niczyjej „winy”. Jeśli chromosomy rodziców są prawidłowe, ryzyko, że choroba powtórzy się u drugiego dziecka jest niewielkie. W przypadku translokacji zrównoważonej u jednego z rodziców, ryzyko wynosi od 9 do 19%.
Na podstawie badań przeprowadzonych we Włoszech stwierdzono, że właściwe badanie rodziny pozwala sprecyzować stopień ryzyka u konkretnej pary. W obydwu przypadkach możliwe jest wykonanie badań prenatalnych. Poradnia genetyczna umożliwia otrzymanie wszystkich niezbędnych do tego celu informacji. Badania chromosomatyczne, a także najnowsze obserwacje z zakresu cytogenetyki molekularnej sugerują obecność tzw. „rejonów krytycznych”, które są odpowiedzialne za typowe oznaki syndromu.
Dwa geny, Semaforyna F (SEMAF – kieruje aksonami i migrującymi prekursorami neuronowymi podczas rozwoju korowego ) i δ -katenina ( CTNND2 – białko zaangażowane w ruchliwość komórek i ulega ekspresji we wczesnym stadium rozwoju neuronów, odpowiada za upośledzenie umysłowe, upośledzenie funkcji poznawczych), które zostały zmapowane do „regionów krytycznych”, są potencjalnie zaangażowane w rozwój mózgu, a ich usunięcie może być związane z opóźnieniem umysłowym u pacjentów z CdCS. Delecja odwrotnej transkryptazy telomerazy ( hTERT – jest czynnikiem ograniczającym szybkość działania telomerazy, niezbędnym do utrzymania długości telomerów i przedłużonej proliferacji komórek), zlokalizowany w 5p15.33, może przyczynić się do zmian fenotypowych w CdCS. Krytyczne regiony zostały niedawno udoskonalone przy użyciu porównawczej hybrydyzacji genomowej macierzy. Krytyczny region przypominający kocie płacz został dodatkowo zawężony za pomocą ilościowej reakcji łańcuchowej polimerazy (PCR) i scharakteryzowano w tym regionie trzy geny kandydujące. Diagnoza opiera się na typowych objawach klinicznych. Analiza kariotypu oraz w przypadkach wątpliwych analiza FISH potwierdzi rozpoznanie. Nie ma swoistej terapii dla CdCS, ale wczesne interwencje rehabilitacyjne i edukacyjne poprawiają rokowanie i poczyniono znaczne postępy w przystosowaniu społecznym pacjentów z CdCS.
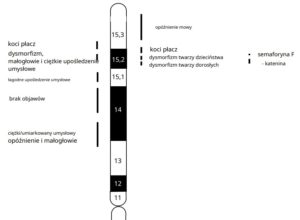
Do tej pory dane na temat powiązania genotypu i fenotypu były nikłe i w wielu kwestiach sprzeczne. Badania przeprowadzone na dużej ilości włoskich dzieci, potwierdziły, że istnieje zmienność kliniczna i cytogenetyczna oraz ukazały powiązanie między zaawansowaniem choroby a typem i rozległością delecji. Rezultaty te mają istotne odzwierciedlenie w praktyce, ponieważ umożliwiają bardziej precyzyjną klasyfikację diagnostyczną poszczególnych dzieci, przydatną w późniejszej rehabilitacji.
Źródło:
Artykuł: Prof. dr hab. med. Alina T. Midro i lek med. Renata Posmyk, Zakład Genetyki Klinicznej, Akademii Medycznej w Białymstoku – Stowarzyszenie na Rzecz Dzieci z Zaburzeniami Genetycznymi GEN
Badania rok 2005: – Mapowanie w wysokiej rozdzielczości relacji genotyp-fenotyp w zespole Cri du Chat przy użyciu porównawczej hybrydyzacji genomowej z macierzą (pobierz pdf: wersja w j.polskim-tłumaczenie google translate, wersja oryginalna w j.angielskim ): Xiaoxiao Zhang, Antoine Snijders, Richard Segraves, Xiuqing Zhang, Anita Niebuhr, Donna Albertson, Huanming Yang, Joe Gray, Erik Niebuhr, Lars Bolund, Dan Pinkel, – High-Resolution Mapping of Genotype-Phenotype Relationships in Cri du Chat Syndrome Using Array Comparative Genomic Hybridization, The American Journal of Human Genetics, Volume 76, Issue 2, 2005, Pages 312-326, ISSN 0002-9297, https://doi.org/10.1086/427762.
(https://www.sciencedirect.com/science/article/pii/S0002929707625828) (